
Making Lenti
The science behind LV Edge, our suite of cells and tools to streamline lentivirus manufacturing. Part II in our series on Engineered Cell Therapies.

It’s Time to Scale
The F.D.A. has approved dozens of cell therapies, clinical interventions that involve placing cells into a person’s body to treat a disease. Nearly half of these approved cell therapies are engineered, which means that the cells are altered to carry added transgenes; a chimeric antigen receptor added to T-cells to treat cancer (Kymriah®), perhaps, or a hemoglobin gene added to blood-making stem cells to treat sickle-cell disease (Lyfgenia®).
Engineered cell therapies are typically made by packaging the transgene into a lentivirus — a type of retrovirus — and then using that lentivirus to deliver the transgene into a patient’s cells. The engineered cells, thus altered, are reinfused back into the body. But there’s a problem: It’s costly and complicated to make lentivirus at scale, and we will need to get better at making these viruses if we stand any chance of making cell therapies more widely available, for more diseases, at a lower cost.
A typical CAR-T therapy costs anywhere from $375,000 and $1,000,000 for a single infusion. This fluctuates from one hospital to the next, of course, but it’s an eye-watering price tag. Biotechnology can bring this cost down, in part by making it easier to make the lentiviruses that are used to make cell therapies in the first place.
Earlier this month, we announced our LV Edge system. It’s an entire suite of tools, including cell lines and software, to help streamline and lower the costs of lentivirus manufacturing. We think it will help the cell therapy space scale up, and also make it simpler for startups and smaller companies to compete.
Bottlenecks
First, a quick recap of how lentiviruses are made, and the bottlenecks we’re trying to solve. (If you read part I, you can skip over this and move to the next section.)
Four plasmids, or loops of DNA, are typically used to make a lentivirus. Each plasmid carries genes that encode different parts of the virus. The genes are split across plasmids to reduce the likelihood that they will recombine to form a replicating, infectious virus. The env plasmid encodes glycoproteins, which sit in the virus’ outer envelope and help it target a patient’s cells. The gag-pol and rev plasmids encode retrovirus structural proteins and enzymes like reverse transcriptase, which converts the lentivirus’ RNA payload into DNA before integration into the patient cells’ genome. The fourth plasmid encodes the transgene of interest; a CAR for cancer therapies, a hemoglobin gene for blood therapies, and so on.
When scientists want to make lentivirus, they often start by taking the well-studied human cell line HEK293, immortalized cells derived from a female fetus in 1973, and transfecting them with the four plasmids. Transfection is just a fancy way of saying that the plasmids are inserted into the HEK293 cells, using either chemicals or electricity. Cells that take up the four plasmids begin to make lentiviruses, which emerge from the cells by cloaking themselves in lipids and budding away into the extracellular environment.
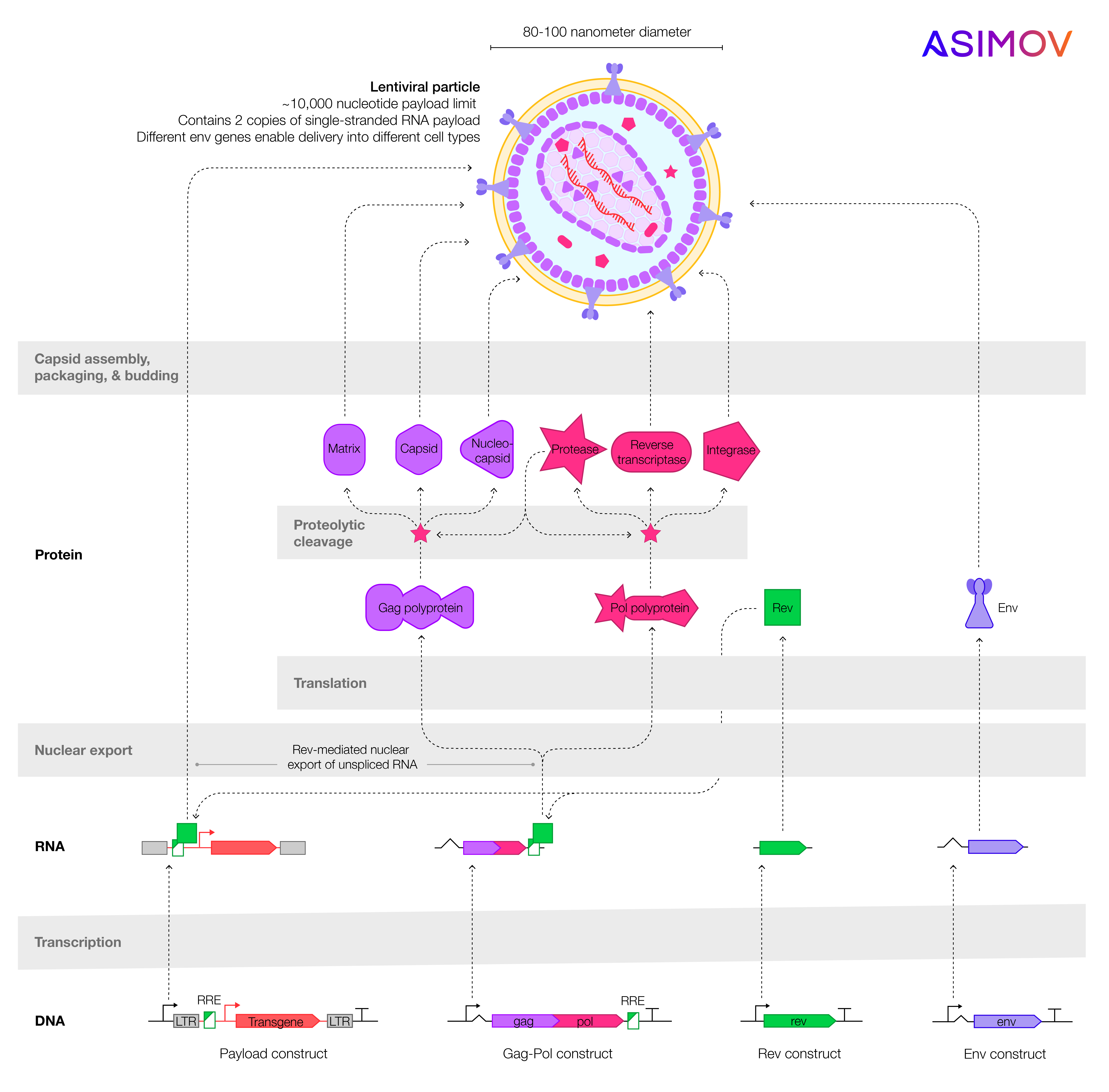
Those are the basics. Now here are the problems.
If a lentivirus is to be used in an F.D.A.-approved therapy, then the plasmids are considered a ‘raw material’ to make that therapy and must be certified as GMP-grade. This certification ensures that the plasmids are safe, pure, effective, and traceable.
But GMP-grade plasmids are inordinately expensive. They can cost hundreds of thousands of dollars per batch, and it can take anywhere from six months to a year to receive them after ordering. Only a few companies make GMP-grade plasmids, so this is a major bottleneck in lentivirus manufacturing.
A second problem is the limitations of transient transfection. When a scientist makes lentiviruses at clinical or commercial scale, they grow normal HEK293 or related cells in a large bioreactor; anywhere from 50 to 1,000 liters in volume. The scientist then floods the cells with the four plasmids, mixing them into the bioreactor. These plasmids enter the cells and coax them to make lentiviruses. The transfected cells die, and the lentiviruses are isolated and purified.
This process is crude. The plasmids don’t always mix evenly in the bioreactor (especially at larger volumes), not every cell receives an equal number of plasmids, and the large bioreactor means that many grams of DNA are required. This manufacturing process has poor scalability and high costs.
Fortunately, both problems can be resolved with advanced genetic design. Our LV Edge system includes a HEK293-derived cell line that already has all the lentivirus genes stably integrated into its genome, and a sequence optimizer tool to boost the expression of lentivirus payloads. But getting these tools to work just right was a tricky engineering challenge.
Engineered Cells
Asimov’s mission is to advance humanity’s ability to design living systems, making it easier to build biotechnologies with outsized societal benefits. We do this by developing four core technology “stacks”: Engineered host cells, genetic systems, CAD software for genetic design, and data-driven models.
The LV Edge system takes advantage of technology we’ve already developed for other applications, such as for therapeutic antibodies. It includes a HEK293-derived host cell line that makes the lentiviruses, and a sequence optimizer that modifies the transgene — or lentivirus “payload,” be it a CAR or something else — to boost expression.
Our engineered cells are called the “packaging line,” because they physically package and make lentiviruses. The packaging cells are HEK293 cells that have all three of the viral plasmids — env, gag-pol, and rev — stably integrated into their genome under inducible control. This solves a key bottleneck in the current way that lentiviruses are made; instead of growing cells in a large bioreactor and then transfecting them with all four plasmids at once, a company using our LV Edge system would only need to transfect one plasmid encoding their transgene of interest. GMP-grade plasmids are still required, but the overall cost of manufacturing is lower.
Soon, we’ll also roll out a “producer line” cell line. These are HEK293 cells that have all of the viral plasmids and the transgene stably integrated into the genome. These cells will fully eliminate the need for companies to transfect cells with GMP-grade plasmids to make lentivirus. It takes a few months for us to integrate a transgene, so the advantage of the packaging line is that it’s more flexible and enables faster timelines.
We’re excited about these cell lines because they remove one of the barriers that make it difficult for the cell therapy sector to scale.
In 1986, the F.D.A. approved the first “recombinant biotherapeutic protein” made by CHO cells. Now, CHO cells make about 70 percent of all biologics drugs sold on the market, including bestsellers like Humira®, whose sales exceeded $21 billion in 2022, and Keytruda®, a cancer therapy with $20.9 billion sold in the same year. Biologics drugs are so popular that, to treat all the patients who need them, the drug industry manufactures many metric tons of these molecules every year. Genentech alone makes 10 metric tons of biologic drugs each year, about 20 percent of the global total.
If cell therapies ever become even a fraction as popular as biologics, then we’ll need to scale manufacturing by a lot. And we think that the only way to reach expected demands in the next decade will be to use stable cell lines and get rid of plasmid transfections entirely.
We had to overcome three challenges to make our cell lines: Stability, titers, and leakiness.
First, stability. Both of our cell lines — the “packaging” and “producer” — were made with an engineered transposase. A transposase is an enzyme that “cuts” and “pastes” chunks of DNA into semi-random sites in a cell’s genome. They’re often used to genomically integrate plasmid DNA. Our engineered transposase has a high efficiency, such that we can stably insert multiple plasmids into HEK293 cells in a single experiment. ‘Stably’ means that the engineered system remains functional over the course of dozens of cell divisions, which matches the number of times a cell will divide while growing in a bioreactor. When a system is unstable, it becomes less and less functional over time, often by epigenetic silencing of the integrated plasmids. After transfection and integration, we screen hundreds of clones and check the top ones to ensure they remain stable over dozens of generations.
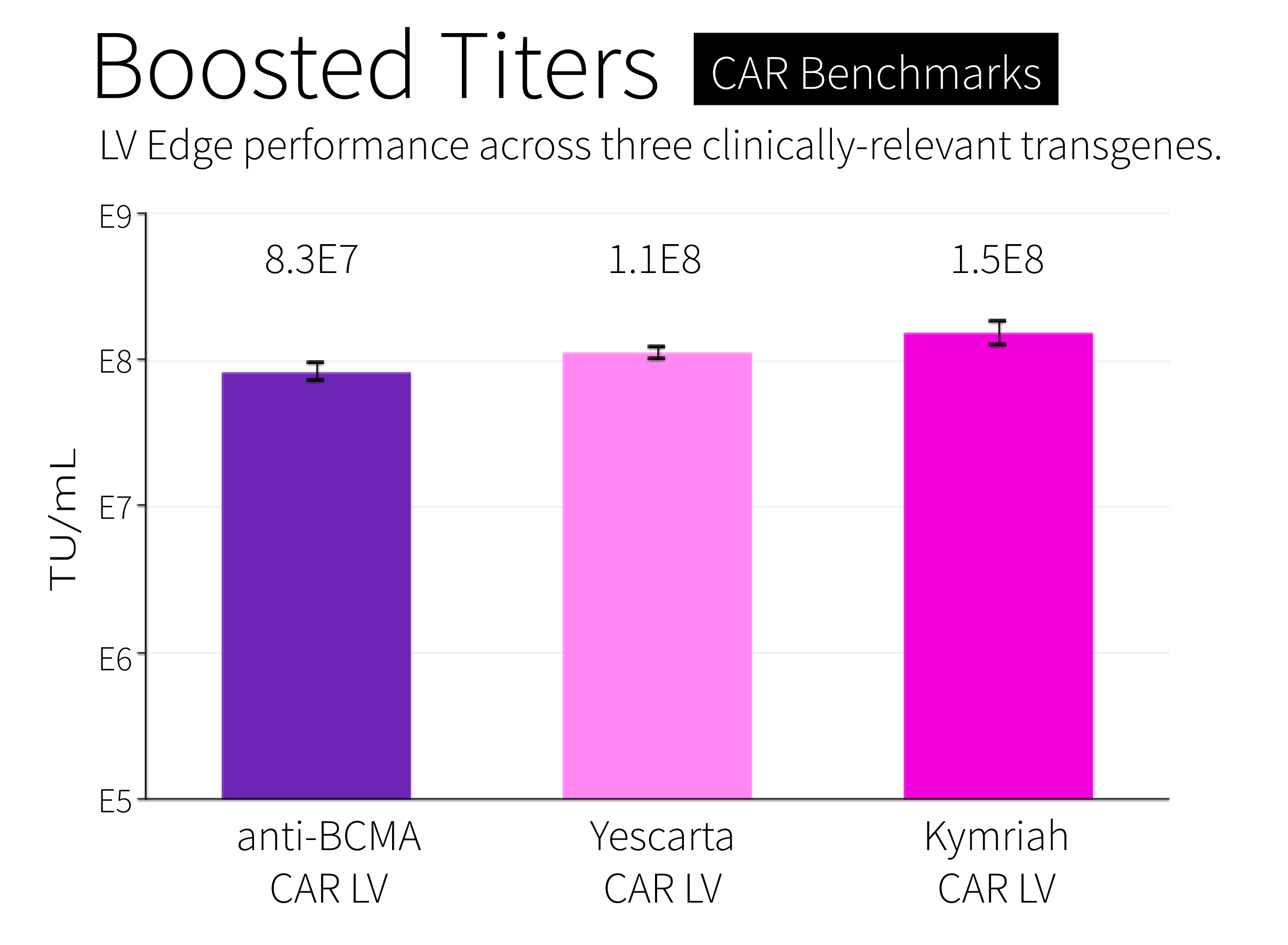
Second, titers. One of the main concerns with doing a stable integration — rather than transient transfections — is that the latter can introduce many more copies of the plasmids into cells, and thus enable HEK293 cells to make a larger amount of active lentivirus. A good titer in the industry is around 1E8 TU/mL before purification, meaning that every milliliter of HEK293 cells makes about 100 million “transducing” units, or viruses.
But there is no point in using a stable cell line, and saving money on GMP-grade plasmids, if the manufacturing titer goes down a hundred-fold to, say, 1E6 TU/mL. That kind of drop in production would force companies to use one hundred-times the bioreactor volume to produce the same amount of lentivirus. That’s not sustainable.
Fortunately, our “packaging” and “producer” cell lines both have titers of 1E8 TU/mL or higher. There is no drop-off in production compared to transient transfections. We tested titers across several transgenes, including the CAR used in Kymriah, and achieved titers of 1.5E8 TU/mL with our “packaging” cell line after sequence optimization (more on that below).
And third, leakiness. Remember how we said that as soon as you put the lentivirus genes into a cell, the cells begin to make viruses and die from the effort? Well, this is a major problem if you’ve integrated all the lentivirus genes into the HEK293 genome, as we have. It’s important to make sure that lentiviruses only get made if cells are first induced with a small molecule, because any “leakiness” of the lentivirus genes could kill the cells. Striking an appropriate balance between zero leakiness and high production was really tricky.
We ultimately figured it out by using a few genetic design and process development tricks. First we made the system controllable by replacing all the viral gene promoters (which initiate RNA transcription) with characterized promoters that can be triggered by an external molecule, without much leak in its absence. Next, we had to shuffle genes around onto different plasmids and make the entire system "integrate-able". Then we cleaned up the system by removing viral elements that may cause uncontrolled plasmid replication. And lastly, we reduced aberrant RNA splicing in our plasmids using transcriptomics data to guide our debugging.
Computational Tools
The LV Edge system also includes computational tools to optimize the lentivirus payload sequence. Kernel is our drag-and-drop, browser-based genetic design software. With Kernel, you can search through hundreds of thousands of genetic parts and insert them onto a canvas to design genetic systems. Kernel also has lab notebooks, an AI assistant, and built-in sequence optimization tools that we’ve developed in-house.
At first, when we tested our cell lines’ ability make lentiviruses packed with various CAR transgenes, including Kymriah, anti-BCMA (used to treat multiple myeloma) and Yescarta (used to treat B-cell lymphoma), we noticed that the titer for Yescarta was substantially lower than the other CARs.
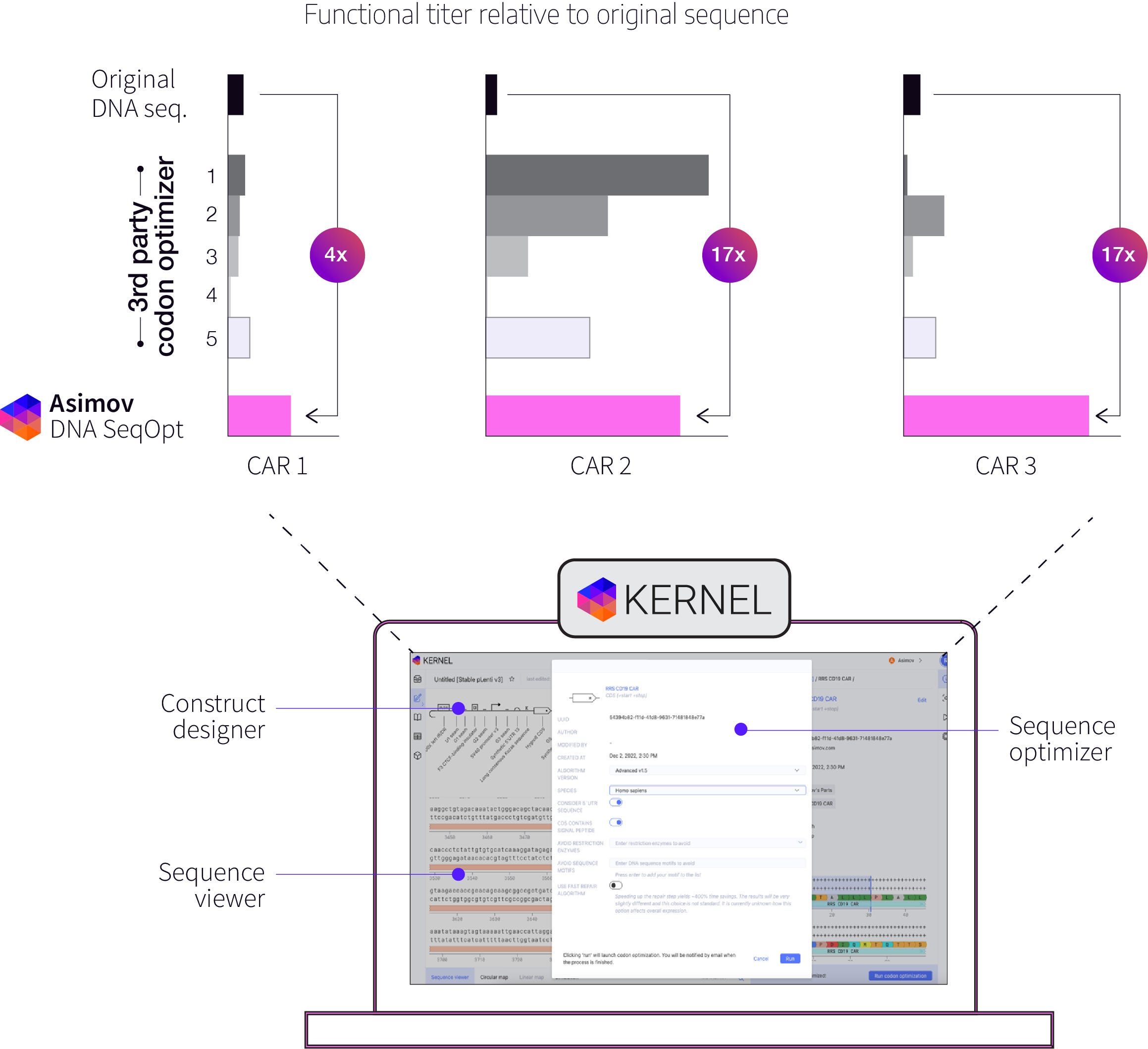
We had previously developed a sequence optimization tool to increase the expression of transgenes, and we tested it out on Yescarta, keeping its amino acid sequence unchanged. Surprisingly, not only did this increase Yescarta’s expression in recipient cells as a payload, it also boosted the lentiviral titer by several fold. This was unexpected; optimizing the sequence of the payload appeared to cause cells to make more packaged virus.
We see this effect for other CARs as well, but we don’t yet fully understand why this happens. Why would optimizing the payload, but not the viral genes involved in packaging, cause a cell to make more virus? We have some hypotheses and we’re running more experiments to better our understanding. In the meantime, we offer our sequence optimization tool to our partners as an additional tool in the toolkit to improve lentivirus manufacturing.
If you’d like to learn more, we’ll be at Advanced Therapies Congress in London in March, at ASGCT in Baltimore in May, and at ESACT in Edinburgh in June. You can also watch our upcoming webinar or visit our website to learn more about our lentivirus engineering efforts.
***
We’re hiring for computational roles. Come work with us! Or subscribe for updates.
Contributors: Jeremy Gam, Alec Nielsen, Mark Stockdale, Michelle Chang, Brianna Jayanthi, Ben Gordon, Jamie Freeman, Raja Srinivas, Chris Thorne, Martín Cárcamo. Words by Niko McCarty.