In 1978, Genentech produced human insulin using engineered E. coli. The achievement signaled a paradigm shift in biotechnology. In the decades that followed, living cells were engineered to produce a suite of medicines, materials, and molecules. Vaccines, cancer gene therapies, synthetic spider silk, and herbicide-resistant plants are all the indelible fruits of this initial biotechnology revolution.
In the last few decades, new tools have lowered biotechnology’s barriers to entry. It has never been easier to work with DNA and engineer living cells. The sheer number of tools developed each year – CRISPR-Cas proteins and AI tools to design genes or proteins are just two examples – have made it relatively simple to manipulate cells. And yet, despite progress, using tools to manipulate and assemble DNA in original and sophisticated ways is still difficult.
At Asimov, we are building a full-stack platform for genetic design, including software to design and ‘debug’ genetic systems, biophysical models to simulate cell processes, and tools to engineer living cells and measure molecules. Our goal is to make biology a true engineering discipline. We want biological engineering to be as robust, reliable, and accessible as electrical, software, or mechanical engineering. If we’re successful, the future will be brighter for all: Cheaper personalized medicines, more bountiful food, and self-healing materials.
At iGEM, you are an integral part of this vision. For many, participation in this global competition is the first step in mastering the hands-on skills required to build a vibrant future. Many of us at Asimov also got our start in iGEM. It’s where we first shared the thrill of engineering biology, while encountering relentless frustrations: Genetic parts that don’t work, plasmids that break, and cells that die for unknown reasons.
We wish we had a guide like this when we were new to synthetic biology. It covers the basics of mammalian genetic design and explains how individual genetic ‘parts’ can be used to build complex transcription units in living cells. This includes everything from promoters and Kozak sequences to basic cloning techniques and cell transfection; all the basic details that you’ll need to program mammalian cells like the human embryonic kidney (HEK), Chinese Hamster Ovary (CHO), and Vero cell-lines used to produce viral vectors, therapeutic antibodies, and vaccine components.
Beyond this guide, Asimov is also engaging with iGEM teams in other ways. For instance, we manufactured the iGEM Distribution Kits, which include all the genetic parts that your team needs to get up and running with experiments. We’re also offering a grant; for select teams, we will synthesize and characterize genetic parts at our state-of-the-art facility in Boston. In addition, all iGEM teams will receive access to our brand-new genetic design tool, called Kernel, which supercharges the genetic design of living organisms. Kernel runs in the browser and can be used to design genetic programs, simply by dragging-and-dropping individual genetic parts in the browser. The software includes a large database, with hundreds of thousands of parts, and these designs can easily be shared within, or across, teams.
We’re excited to support you this year, and honored to be a part of iGEM. Come learn more about our work at Asimov.com.
– The team at Asimov
Living cells communicate, sense, and respond to their neighbors and environment. White blood cells hunt down pathogens inside the body by following trace amounts of chemicals secreted by the microbes. Dense communities of microbes in the wild, or in your gut, share food and distribute tasks between members.
These behaviors appear distinct, but are governed by a common principle: Cellular functions arise from the expression of their genes.
For a synthetic biologist to harness nature’s potential, and access its machinery to build molecular medicines, or pattern a material akin to a leopard’s spots, or break down harmful pollutants, they must first understand how cells process the world around them, and then precisely alter genetic material, within those cells, to program new biological functions.
That second bit – alter genetic material – sounds somewhat simple. After all, there are many tools to genetically modify cells, and it has never been cheaper and easier to write new DNA sequences or edit genes in a genome. Still, the challenge remains: What to write? Or, put another way: How are design concepts best converted into DNA sequences and genome alterations? Moreover, it remains challenging to alter living cells in a manner that is both reproducible and predictable. Without design guidance, even a simple task, like engineering a cell to sense a pollutant and switch on a green fluorescent protein signal in response, is typically done ad hoc, and is therefore prone to failure or unreliable performance.
This Guide will teach you to move beyond bespoke methods and employ best practices to program advanced behaviors in mammalian cells in a predictable manner. It will teach you how genetic parts, which include promoters and coding sequences, can be stitched together to assemble a complete transcription unit. It also explains how DNA can be modified and rearranged to enable new behaviors, and how collecting a lot of data on individual sequences enables one to program biology more reliably. In other words, it explains the fundamentals of mammalian genetic design, which is the science of altering the DNA of a cell in order to change how it functions. Genetic design uses advanced biological techniques and data-driven computational approaches, including characterized DNA sequences, modeling, and multi-omics analysis, to achieve those aims.
This Guide is explicitly focused on mammalian cells, rather than bacteria. Many biotechnology companies use CHO or HEK cells to manufacture antibodies or medicines, so learning to work with these cell lines will prove valuable long after your iGEM project is complete.
In the next section, we dive into the principles of genetic design by walking through the Central Dogma, the fundamental biological process that guides all biological engineering.
Basics of Genetic Design
Living cells encode complex networks of genes that work together, in a brilliant symphony, to control cell division, metabolism, movement, and everything else. The human genome alone encodes about 20,000 genes. Before one can engineer a living cell to sense a signal and turn on a gene’s expression, it’s important to first understand how cells normally carry out these functions. Gene expression is governed by the Central Dogma, which defines how a genetic sequence is converted into a protein (read our blog post The Central Dogma in 7 Experiments here).
The Central Dogma, in mammalian cells, works like this:
First, DNA is transcribed into RNA by a large protein complex, called RNA polymerase. Mammalian cells contain three types of RNA polymerases; type I, II, and III. RNA polymerase I synthesizes about 60 percent of all cellular RNAs, and is also responsible for transcribing the RNA strands that make up the core of ribosomes (which are proteins that build other proteins). RNA polymerase II, widely considered to be the “canonical” RNA polymerase, converts DNA into messenger RNA, and also synthesizes small nuclear RNAs and microRNAs. RNA polymerase III synthesizes tRNAs and other RNAs, which help ribosomes assemble proteins.
Let’s focus strictly on RNA polymerase II. To carry out transcription, this protein first latches onto a DNA sequence called a promoter, which contains a binding site for the protein complex. RNA polymerase II flies down the DNA strand and transcribes about 40 nucleotides into mRNA per second. In mammalian cells, this process occurs within the cell nucleus.
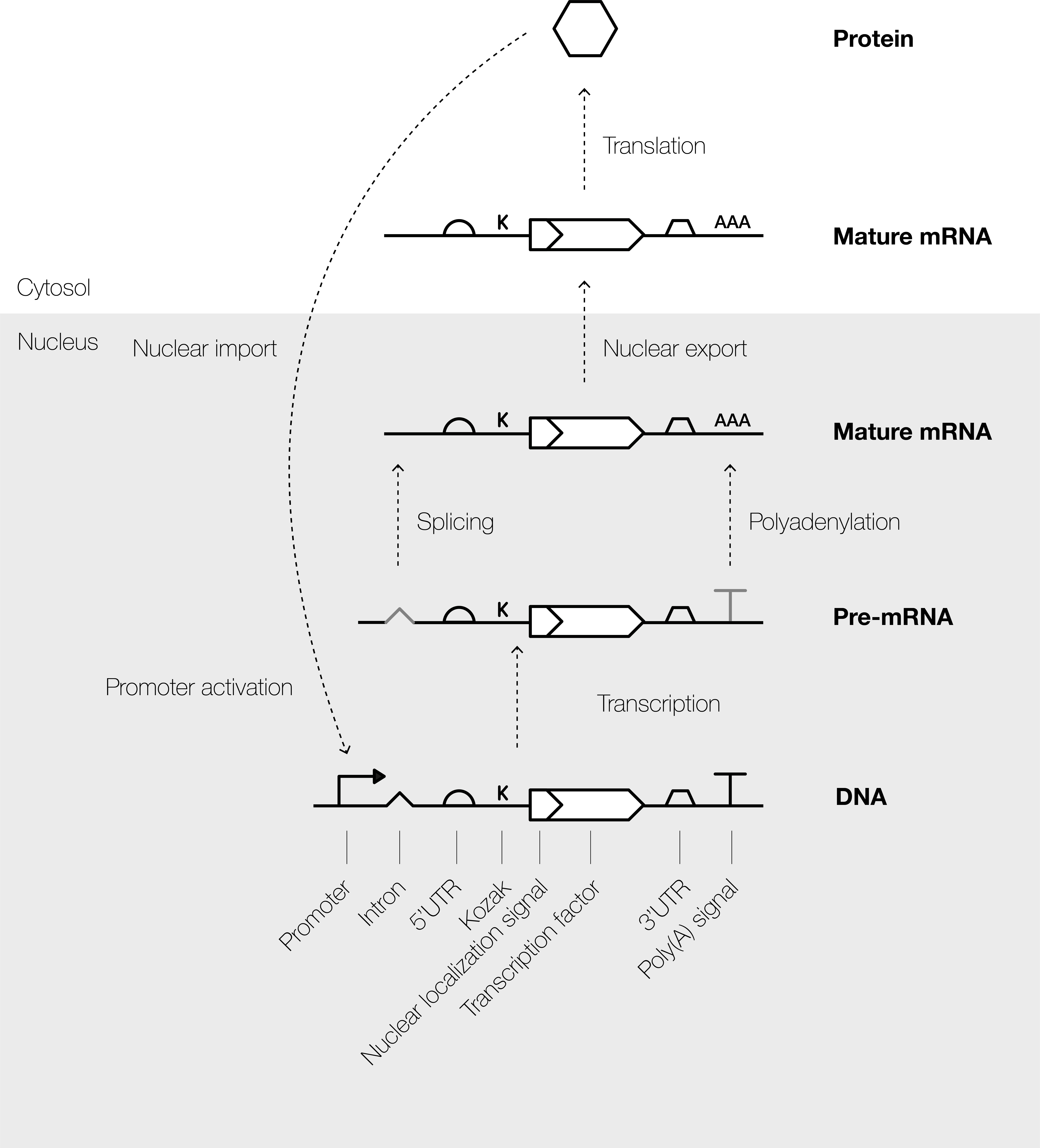
After transcription, a nascent mRNA strand is produced and is quickly processed to form a “mature” mRNA. During mRNA maturation, a special ‘cap’ nucleotide is added to the 5’ end of the mRNA, and introns – sequences within the mRNA strand that don’t encode a protein – are removed. Toward the tail end of the mRNA transcript, enzymes within the cell’s nucleus recognize a poly(A) signal sequence, cleaving the strand and adding a long chain of adenine nucleotides – a so-called poly(A) tail – to the 3’ end of the mRNA strand. This tail protects mRNA from degradation. A typical poly(A) tail contains anywhere from 100 to 250 adenines in a row. The mature mRNA strand moves through the nuclear pore complex and into the cytoplasm.
The next step is translation. This process begins when a ribosome latches onto the mRNA’s ‘cap,’ scans along, and then strikes upon the Kozak sequence, the site of translation initiation. In mammalian cells, the Kozak sequence includes the “start” codon of the protein coding sequence. (In bacteria, there is an analogous sequence for translation initiation, called a ribosome binding site.)
After initiating translation, ribosomes ratchet along an RNA sequence and decode three nucleotides at a time. Each of these so-called triplets (a “codon”) within a coding sequence, or CDS, encodes a single amino acid, which the ribosome appends to a growing polypeptide chain. The ribosome adds about five amino acids per second to a polypeptide in human cells. At the end of this process, when the ribosome reaches a stop codon (UAA, UGA, or UAG), a fully-formed protein is released.
That’s the Central Dogma in a nutshell: DNA is transcribed to RNA, which is translated to protein.
Now let’s briefly consider how one could “play” with these rules to engineer living cells that have entirely new functions. To begin, note the several types of DNA sequences above that influence or control the execution of the central dogma (e.g. promoters, Kozak sequences, coding sequences, and stop codons, so far). Knowing this, one could swap one sequence for another, such as by dropping in a new coding sequence, to “trick” a ribosome into making a new type of protein. It is also possible to take a gene from one organism, such as a daffodil, and put it into another organism, such as rice. Researchers did exactly that two decades ago to create engineered rice that biosynthesize beta-carotene, a vitamin A precursor that could help save the lives and protect the eyesight of millions of children in Africa and Southeast Asia.
Apart from swapping a coding sequence, one could also swap out promoters to increase or decrease the number of mRNAs that are transcribed from a single gene. In fact, each type of DNA sequence “part” that makes up a standard gene is interchangeable. These parts can be rearranged or modified, as needed, to construct transcription units with desired properties.
A typical transcription unit, in mammalian cells, contains the parts described above, as well as some new ones. These are the promoter, 5’ untranslated region (UTR), Kozak sequence, coding sequence (CDS), 3'UTR, and poly(A) signal (which is distinct from the poly(A)-tail).
Pictorially, a transcription unit is represented like this:

In the next section, we’ll go on a tour of the various parts that make up a transcription unit, and explain how you might use them to build genetic designs in mammalian cells.
But first, a caveat: It may seem relatively simple to build a transcription unit – just swap parts around! Yippee! – but biology is a lot more difficult than that. In living cells, there are exceptions to every rule. When one tries to arrange genetic pieces in new ways, the genetic designs often fail, and we don’t always know why.
Although we conceptualize genetic parts as simply “promoters” or “poly(A) signals,” it’s important to remember that each part is a physical DNA sequence. Cells do not care about our abstractions; they only see As, Ts, Cs, and Gs. If we make a mistake while building a genetic system, it’s possible to unintentionally introduce a ‘hidden’ promoter or other sequences that send the wrong message to the cell.
Sometimes, the genes we introduce are also toxic, or certain promoters coax cells to produce so much RNA that the organism becomes overwhelmed by the sheer amount of metabolic resources required, capitulates, and dies. One must be careful to balance the expression of each transcription unit to make enough protein while simultaneously preserving the cell’s health.
In the next part of this guide, we explain how each part works, at a basic level, and how to use them to build genetic designs with desired behaviors. We give special emphasis to mammalian genetic parts of the types that are included in this year’s iGEM Distribution Kit. Although there are many analogous parts in microbial cells, there are also important differences. Later in this guide, we explain how to stitch together genetic parts to build transcription units and how to transfect them into mammalian cells.
Individual Parts
The iGEM Distribution Kit includes the new Mammalian Parts Collection, which has everything you need to get up and running with mammalian genetic engineering. The Mammalian Parts Collection includes 21 single parts that can be used to assemble new transcriptional units, as well as several ‘empty’ plasmids that can be used to clone new parts. The Collection also includes a series of positive controls, which will prove useful when you begin transfecting cells and troubleshooting experiments.
Each of the following sections are focused on mammalian cells. Any time a biological process is described, one should assume we are referring to mammalian cells, unless otherwise stated.
Now let’s dive in!
Promoters

Promoters are the starting point for exerting control over a gene’s expression. Promoters can be designed to modulate the level of RNAs and proteins in a cell or, in some cases, be used to trigger gene expression only after specific events occur.
Inside a cell, promoters are the site where transcription factors and RNA polymerase initiate transcription. Promoters are bound first by transcription factors, which then kick off a cascade of molecular events that bring RNA polymerase to the site.
Promoters are situated upstream of 5'UTRs and coding sequences. Both prokaryotes and eukaryotes have promoters, which can be either constitutive or inducible. The former indicates that a promoter is always “on” and transcriptionally active, while the latter indicates that a promoter can be switched “on” or “off” with an inducer, typically a chemical or other signal, such as light.
Mammalian promoter sequences are typically between 100 and 1,500 base pairs in length. Many are made up of transcription factor binding sites, a TATA box, an Initiator element, and a ‘core’ promoter element. The first transcribed nucleotide in a gene is denoted with an index of +1, while nucleotides upstream of a transcription start site are denoted with negative numbers (there is no index 0!)
Details: When people talk about promoters, you may hear terms like “strong” or “weak.” A strong promoter produces more copies of mRNA per DNA template, as it recruits RNA polymerase more strongly. More mRNA is often, but not always, associated with higher protein levels.
Recall that mammalian cells contain three different types of RNA polymerase proteins; I, II, and III. Each type of RNA polymerase recognizes a unique promoter sequence. For most synthetic biology applications — or any situation in which a gene is intended to produce a final protein — one should use Pol II promoters. To express a CRISPR guide RNA or other non protein-coding RNA inside of a cell, one should typically use a Pol III promoter instead. Pol I promoters are devoted, in mammalian cells, to the transcription of ribosomal RNA genes.
Recall, also, that promoter sequences can be designed to switch on or off in the presence of a molecule, shift in temperature, or specific wavelength of light. These promoters are called inducible, and several of them are commonly used in synthetic biology research, including the Tet-On and Tet-Off systems. These parts are not included in the iGEM Collection, but their DNA sequences are publicly available online. Typically, an inducible promoter requires an additional transcription factor to be introduced into the cell for it to function.
Tet-Off is an engineered transcription factor that activates transcription from its cognate promoter in the absence of a small molecule inducer, called doxycycline. The transcription activator protein is actually a fusion of two proteins: TetR (a DNA-binding protein from the bacterium E. coli) and VP16 (an activation domain from the herpes simplex virus). The Tet-On system works in the opposite way; transcription activation occurs when doxycycline is added.
Inducible promoters are sometimes “leaky,” which means that they are somewhat transcriptionally active even when in an “Off” state. Leakiness is normal, but it can be reduced by combining multiple inducible systems so that a gene is only switched on if multiple inducers are added at the same time.
There are also regulatable promoters, which enable fine-tuned control over gene expression; they can be activated or repressed by specific proteins.
Note: Avoid encoding separate genes, with separate promoters, in convergent orientations. This can cause transcriptional interference and mess up your designs, reducing the expression of both genes. It’s typically best to encode genes so that they point in the same direction (tandem orientation), or face away from one another (divergent orientation). Additionally, certain promoters (such as the CMV promoter and its variants) are prone to epigenetic silencing over time, which means that their expression levels gradually dwindle in mammalian cells. Avoid using these promoters in designs that require long-term gene expression.

What’s In the Collection? The Mammalian Parts Collection includes three constitutive promoters and one regulatable promoter. The promoters are called hef1a, CMV, and EFS. The Collection also includes a regulatable UAS-Gal4-minCMV promoter; its expression is normally low, but ramps up in the presence of a protein called Gal4.
The Collection does not contain promoters for type-I or type-III RNA polymerases, which are typically used to express RNAs that should not encode proteins, such as a CRISPR-Cas guide RNA, a tRNA, or small RNAs for RNAi-based gene silencing. The sequence for a common Pol III promoter, called U6, is publicly available online.
5'UTR and Kozak Sequence

A 5'UTR and Kozak sequence are the levers for increasing or decreasing the ribosome’s rate of protein translation from an mRNA. Tuning these sequences can be used to control protein levels in the cell.
Just to refresh, a 5'UTR is the untranslated nucleotide sequence that stretches upstream from the start codon. In an mRNA strand, it’s the sequence that is bound and scanned by ribosomes before initiating protein translation. 5’UTRs control the translation initiation rate in many ways:
First, a 5’UTR sequence can cause an mRNA to fold up on itself in a way that makes it difficult for the ribosome to bind, or recognize, the start of its CDS. A 5’UTR may also encode upstream open reading frames, or uORFs, which are short protein coding sequences that effectively “use up” the ribosome before it gets to the downstream CDS of interest. Additionally, some 5'UTRs include introns, which are non-coding sequences that are spliced out of an RNA strand and tend to increase the translation rate.
Besides influencing how often ribosomes initiate translation of the CDS, these sequences also dictate the half-life of the mRNA, or how long the strand persists before being degraded by the cell. This, in turn, influences how many proteins can be translated from an mRNA. All of these details are important to understand when designing genetic systems that must behave reliably, over extended time periods.
Mammalian cells also have a short RNA motif, called a Kozak sequence, that overlaps with the end of the 5'UTR and the beginning of the coding sequence. A pioneering biochemist named Marilyn Kozak discovered, in the 1980s, that the 6 nucleotides before the initiation codon, as well as the nucleotide that immediately follows, strongly influence translation rates.
The Kozak sequence is not fixed in stone, either; it can be altered to change the number of proteins produced from an mRNA.

Details: A 5'UTR can be anywhere from tens to thousands of nucleotides in length. Changing the sequence of a 5'UTR can more than double the number of proteins produced by a given mRNA sequence.
If designing a 5'UTR from scratch, it is typically best to make it less than 100 base pairs in length, and to ensure that the sequence has a weak RNA secondary structure (RNA structure prediction calculators, such as RNAfold, are available online). To make 5'UTRs that result in a low translation initiation rate of your downstream CDS, place one or more short upstream open reading frames (“uORFs”), containing a start codon and in-frame stop codon, within the 5'UTR.
Note: The 5’UTR technically begins at the transcription start site (the +1 index), which is inside of the promoter. However, in this Guide, we refer to the sequence between the promoter and the CDS as the 5’UTR. In that sense, the 5’UTR parts that we describe are actually 5’UTR segments.
What’s In the Collection? The Mammalian Parts Collection includes four 5'UTR sequences, called 5UTR13, uORFs(1x weak), uORFs(1x) and uORFs(4x). The 5'UTR that will yield the highest protein levels is 5UTR13. The various uORF sequences reduce the amount of protein translated from a gene, with uORFs(4x) reducing translation the most.
Kozak sequences are not provided as a separate part in the Collection because they are already attached to coding sequences, which we discuss next.
Coding Sequences

A coding sequence, or CDS, is the portion of a gene that encodes a protein, which is the main functional unit within living cells. One can design and build entirely new proteins, that exist nowhere in nature, by tinkering with a CDS. Place each CDS before a 3'UTR and poly(A) signal.
Details: A typical human protein contains between 300 and 500 amino acids, which means that most coding sequences are about 1,000 to 1,500 base pairs in length. But this is just an approximation! One gene, called dystrophin, is 2.5 million base pairs long! If you stretched out the dystrophin gene, using a pair of tweezers, it’d measure nearly 1 millimeter. The dystrophin protein links actin to other proteins and plays important roles in muscle contraction. Some tRNAs, in comparison, are only about 200 bases in length.
Now, the important thing to know about a CDS is that they can come from any kingdom of life; from the smallest bacterium to a blue whale or towering tree. Researchers often find CDS’ by sequencing the world around them, and then using computational tools to pinpoint the locations of genes within a sequence.
But if you wanted to take a CDS from a daffodil, for example, and put it into rice, there are some changes you’d have to make first. Recall, from your textbooks, that cells use 20 different amino acids to build proteins. These amino acids are encoded by 61 different codons (there are also three ‘stop’ codons), and some amino acids are encoded by multiple different triplet codons. ‘AUG’ is the de facto start codon, while CUU, CUC, CUA, UUA, UUG, and CUG all encode the same amino acid: leucine.
Every organism has its own ‘preferences’ for codon usage. E. coli bacteria encode 47 percent of all leucine amino acids using the CUG codon, while yeast use that codon for just 11 percent of leucines. The process of tweaking the codons in a CDS, in order to optimize its expression in a new host, is called codon optimization.
Since different organisms have varied preferences for codons, swapping out rare codons for more frequent ones is often a simple way to improve translation in the new host. Many codon optimizer tools are available online, and there are also other, more advanced optimizations that can be done to remove problematic sequence motifs or modulate the folding energy of an RNA molecule.
A CDS sequence can be modified in other ways, too. The first amino acid in a protein is called the N-terminus because it has a free amine group, while the last amino acid is called the C-terminus because it has a free carboxyl group. Multiple protein domains can be encoded, one CDS after the other, by linking them together at their termini to form a single, unbroken polypeptide chain.
For example, signal peptides are small peptide sequences that, when added to a protein’s N-terminus, tell the cell to secrete a protein into the extracellular environment. (Signal peptides are removed from proteins during secretion.) Individual proteins can also be tagged with a nuclear localization signal, thus targeting them to a cell’s nucleus.
Note: Not all coding sequences are created equal. The more G or C nucleotides that a gene sequence contains, the higher the stability of the mRNA sequence (usually). Be cautious when selecting stop codons, too; some are less effective at halting translation.
What’s In the Collection? The Mammalian Parts Collection includes six coding sequences, including four fluorescent proteins, one luciferase protein, and one transcription factor. It includes Gaussia luciferase, mRuby2, TagBFP, YFP, and iRFP720. It also includes Gal4-VP16, a transcription factor that recognizes, and activates transcription from, the UAS-Gal4-minCMV promoter sequence. Every CDS in the Mammalian Parts Collection contains a built-in upstream Kozak sequence; these are not provided as separate parts.
Gaussia luciferase is a small, bioluminescent protein. It is commonly used as a reporter signal to monitor biological processes. This CDS includes a N-terminal signal peptide, and is therefore secreted from the cell.
mRuby2, TagBFP, YFP, and iRFP720 are all fluorescent proteins. These proteins absorb photons of a specific wavelength and emit photons of a longer wavelength. mRuby2, for instance, is typically excited by light with a wavelength around 560 nanometers. A free online database, called FPbase, can be used to check the specific emission and excitation wavelengths for fluorescent proteins.
Gal4-VP16 activates expression of CDS’ placed downstream of the UAS-Gal4-minCMV promoter. It can be used to build inducible transcription units.
3'UTR

A 3’ untranslated region, or 3'UTR, exerts control over the stability and trafficking of an mRNA transcript. It should be placed downstream from a protein coding sequence, but before a poly(A) signal sequence.
Details: A typical 3'UTR sequence can be anywhere from tens of nucleotides to thousands of nucleotides in length. Altering a 3'UTR sequence can have a large effect on the fates of individual mRNAs, too. For one study, researchers plotted the association between 3'UTR sequences, taken from more than 2,000 human genes, and mRNA abundance, stability, and protein production. They found many 3'UTRs that had a large effect on each parameter – some 3'UTRs caused mRNAs to degrade in just one hour, while others persisted for several hours.
What’s In the Collection? The Mammalian Parts Collection includes two 3'UTR sequences. Both of these sequences are synthetic, which means that they don’t appear in natural organisms. The two 3'UTR sequences are called Synthetic 3’UTR v1 and Synthetic 3’UTR v2.
Poly(A) signals

A poly(A) signal is the final piece of a transcriptional unit. It is the RNA sequence that directs mRNAs to be cleaved and then decorated with a chain of adenines (a poly(A) tail). In mammalian cells, poly(A) signals terminate transcription. Here, again, slight changes in a poly(A) sequence can have large ramifications on a gene’s expression levels; they also change how quickly an mRNA sequence decays, “matures,” and moves out of the nucleus.
Details: Poly(A) signals that contain UGUA, followed by either AAUAAA or AUUAAA, seem to be most effective at halting RNA polymerase. It is also possible to place two poly(A) signals, one after the other, to further increase the transcription termination efficiency and reduce the amount of RNA polymerase that transcribes through one transcription unit and into another.
What’s In the Collection? The Mammalian Parts Collection includes two poly(A) sequences. They are called RB glob and bGH. Each of these parts comes from a natural sequence in a mammalian genome; RB glob is derived from the rabbit beta globin gene and bGH is derived from the bovine growth hormone gene.
Other Parts
We’ve covered the five types of parts that are provided in this year’s Mammalian Parts Collection. However, there are additional part types that may be useful in your project.
An enhancer is a regulatory DNA sequence, in mammalian cells, that can be used to boost a gene’s expression levels or control transcription in specific cell types. Enhancers should be placed within a few thousand bases of an associated promoter sequence, either upstream or downstream, and in either a forward or reverse orientation.

An insulator is a DNA sequence that counteracts enhancers. These DNA sequences can be used to “insulate” transcription units from their surrounding genetic contexts. Typically, insulators are used when genetic designs are stably integrated in a chromosome: One placed upstream of the promoter, and a second placed downstream from the poly(A).

An internal ribosome entry site (IRES) can be used to translate multiple independent proteins from one mRNA strand. These sequences initiate translation without requiring that a ribosome scans along after binding to a 5’ mRNA cap. An IRES should be placed upstream from a CDS, but within a transcribed region. There are many IRES variants, and using them to translate two proteins from a single piece of mRNA often leads to varying protein levels (e.g. the first gene is translated more than the second.)
These sequences come from viruses, and they enable synthetic biologists to do something that, at least in bacteria, we often take for granted: Namely, express multiple genes from a single mRNA. Bacteria do this all the time, but mammalian cells don’t; an IRES is a nice tool to build “operons” in the latter.

A 2A tag, or ribosomal skip peptide, is a coding sequence that causes the ribosome to skip one peptide bond during translation, resulting in two non-covalently linked polypeptides from a single open reading frame. 2A tags should be placed in-frame, directly between two protein coding sequences. If using a 2A tag, it’s best not to include a stop codon in the first CDS.
A 2A tag is anywhere from 18-24 amino acids in length, and there are several options to choose from. Just note that, if chaining together multiple proteins with 2A sequences, the first CDS is usually expressed more highly than the last (much like the IRES). 2A sequences have been used to link together at least four proteins.

As you learn about more parts, and how they work, you’ll gradually be able to build more complex genetic systems. One especially complex example is depicted below – we made it using Asimov’s genetic design software, called Kernel, and the sequence includes everything from insulators and 2A tags to enhancers and an IRES.
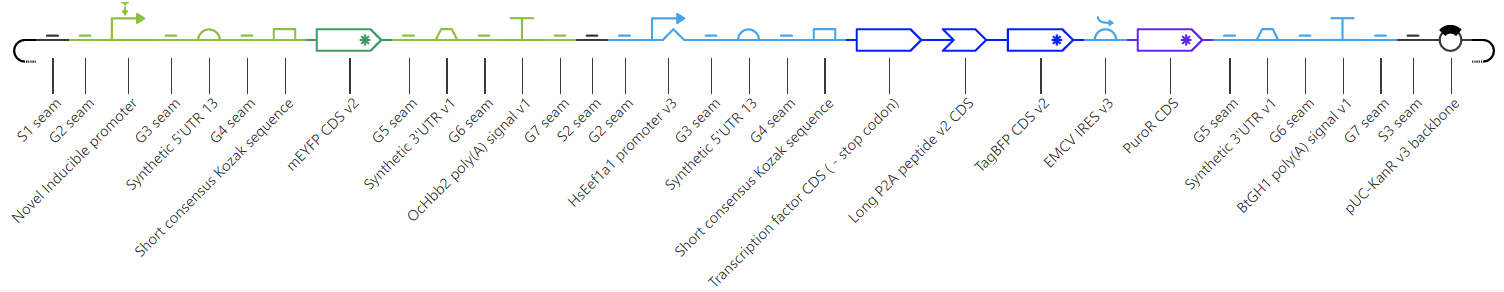
Let’s take a quick look at the design, which shows a genetic circuit that encodes a yellow fluorescent protein, downstream from an inducible promoter. A second promoter drives expression of a transcription factor, which activates the inducible promoter in the presence of a small molecule, and a blue fluorescent protein. The two are separated by a 2A tag, and the presence of BFP is used to visualize how well the transcription factor expresses in mammalian cells. Additionally, an IRES element separates the two CDS' from a coding sequence for a puromycin resistance protein. Puromycin is a type of antibiotic that can be used to select for transfected cells. All of these components are contained in just two transcription units.
How to Make A New Part
This year’s Mammalian Parts Collection in the iGEM Distribution Kit contains many parts that can be used to build transcription units in mammalian cells. A basic transcription unit contains a promoter, 5'UTR, coding sequence, 3'UTR, and poly(A) signal. Each of these parts can be stitched to the others using the Type IIS assembly method. More details about this method can be found on the iGEM website.
At its basic level, Type IIS assembly uses restriction enzymes to cut DNA plasmids containing each type of part, such as promoters and 3'UTRs, and produce unique overhang sequences that are then joined and ligated with other parts. The site at which two parts join is called a “seam.”
There are three “levels” to Type IIS assembly. Level 0 (L0) denotes an individual part, Level 1 (L1) denotes a single transcription unit, and Level 2 (L2) denotes multiple transcription units that have been joined together. Before making a transcription unit (L1), all the necessary parts must first be in a L0 plasmid.
New genetic parts (L0) can be generated by creating a piece of double-stranded DNA (using de novo synthesis, polymerase chain reaction, or any other method) that includes the sequence of interest (such as a promoter, 3'UTR, or poly(A) signal), ‘seams’ matching the pL0 backbone, and restriction enzyme recognition sites for BbsI. The sequence of the part itself should not contain additional BbsI, BsaI, or SapI recognition sites, because these will interfere with the cloning process. If one of these recognition sites already exists within a sequence, it should be removed by making synonymous mutations, where possible.
The BbsI restriction enzyme recognizes:
5’... GAAGAC NN ... 3’
3’... CTTCTG NNNNNN ... 5’
The ‘N’ nucleotides, in this schematic, can include any nucleotide: A, T, G, or C. Restriction digestion with BbsI leaves behind two “gap” nucleotides, as well as a four-base, single-stranded overhang sequence, called a ‘sticky end.’ The overhang sequences should be designed such that a promoter can be attached to a 5'UTR, a 5'UTR attached to a CDS, and so on. More details on Type IIS assembly and restriction enzymes are available on the New England Biolabs website. The overhangs required for each type of part are:
- L0 promoter: GCTT—[Promoter]—CAAC
- L0 5'UTR: CAAC—[5'UTR]—GCAA
- L0 CDS: GCAA—[Kozak-CDS]—CCCA
- L0 3'UTR: CCCA—[3'UTR]—GAGT
- L0 poly(A): GAGT—[Poly(A)]—TACA
Each new part should be cloned into a L0 destination plasmid. The Mammalian Parts Collection includes five L0 destination plasmids; one for each type of basic part. Each L0 destination plasmid encodes a gene for superfolder green fluorescent protein, or sfGFP, enabling these plasmids to be observed under a microscope or with a transilluminator after cloning and transformation into a bacterial host.
When a L0 destination plasmid is cleaved with BbsI, the gene encoding sfGFP is removed and replaced with the new part. Bacterial cells that have a plasmid with the cloned insert will not glow green. The backbones of the L0 destination plasmids also encode a gene that confers bacteria with resistance to antibiotics called ampicillin and carbenicillin. Transformed cells with the plasmid should grow on LB agar plates that contain 100 micrograms per milliliter (µg/mL) of ampicillin or carbenicillin.
Once a new part has been cloned into its L0 destination plasmid, it should be miniprepped and sent for sequencing to ensure that the overhangs and inserted sequence are correct. Colony PCR can also be performed to check the length of the sequence inserted in the plasmid. Several companies now offer long-read sequencing as a service, including Plasmidsaurus and Primordium Labs.
How to Build A Transcription Unit
A transcription unit is a stretch of DNA that begins with a promoter, ends with a poly(A) signal, and includes other elements in-between. A transcription unit can be assembled by piecing together a series of L0 parts. A single transcription unit is denoted as L1.
The process to build a L1 transcription unit is quite similar to the process for building a L0 level part. To make a transcription unit, first collect all of the required L0 components, including a promoter, 5'UTR, Kozak-CDS, 3'UTR, and poly(A) signal. Each L0 plasmid includes restriction sites that are recognized by the enzyme BsaI. This restriction enzyme recognizes:
5’... GGTCTC N ... 3’
3’... CCAGAG NNNNN ... 5’
The ‘N’ nucleotides, in this schematic, can include any nucleotide: A, T, G, or C. DNA digestion with BsaI leaves behind one “gap” nucleotide, as well as a four-base overhang sequence. More details are available on the New England Biolabs website.
Each L0 plasmid should be cleaved with BsaI, in addition to a L1 destination plasmid. All six components should then be ligated together to make a new plasmid that contains a transcription unit with the selected promoter, 3'UTR, and other components. This technique is often called Golden Gate Assembly, and a video explaining the process can be viewed here.
The backbones of each L1 destination plasmid encodes a gene that confers resistance to an antibiotic called spectinomycin. Transformed E. coli cells that received the plasmid via transformation should be able to grow on LB agar plates that contain 50 µg/mL of spectinomycin.
The Mammalian Parts Collection includes four L1 destination plasmids. Each destination plasmid also encodes a gene for lacZ. This gene encodes a protein called β-galactosidase, which enzymatically cleaves lactose sugars into glucose and galactose. Cells that carry a plasmid with the lacZ gene, and which are grown in the presence of both X-gal (galactose molecules linked to substituted indoles) and 1 µM of IPTG will appear as blue colonies. Cells lacking the lacZ gene (which indicates that the gene has been removed and replaced with the transcription unit) will appear white.
Multiple transcription units can also be joined together into multi-transcription unit constructs. The Mammalian Parts Collection includes three L2 destination plasmids. These can be used to make plasmids that contain anywhere from 2 - 4 total transcription units.
To assemble multiple L1 transcription units into a L2 multi-transcription unit construct, cut each L1 plasmid with the SapI restriction enzyme. This restriction enzyme recognizes:
5’... GCTCTTC N ... 3’
3’... CGAGAAG NNNN ... 5’
More details are available on the New England Biolabs website. Again, the ‘N’ nucleotides, in this schematic, can include any nucleotide: A, T, G, or C. DNA digestion with SapI leaves behind one “gap” nucleotide, as well as a three-base overhang sequence.
Each L1 transcription unit, as well as a L2 destination plasmid, should be cleaved with SapI and then ligated together. The backbones of each L2 destination plasmid encodes a gene that confers bacteria with resistance to an antibiotic called kanamycin. Transformed E. coli cells that receive a cloned plasmid via transformation should be able to grow on LB agar plates that contain 50 µg/mL of kanamycin.
Additional details on the Type IIS Assembly Method and Golden Gate cloning are provided in this open access article from Bird J.E. et al. in ACS Synthetic Biology. The iGEM website also includes a page on Type II Assembly, although it is specific to bacterial cells. (Note: Mammalian transcription units are made from five parts, whereas bacterial transcription units are made from four parts. Thus, the overhangs used to piece together parts during Type IIS assembly can vary between organisms.)
Getting DNA into Cells
Bacteria and mammalian cells are quite different from one another. The former can be transformed in a few minutes, with colonies appearing a few hours later. Mammalian cells are more difficult to work with. Simply getting DNA into a mammalian cell requires two days of work. Unlike bacteria, plasmids typically do not replicate inside of mammalian cells (an exception is plasmids with viral origins of replication, but these are not commonly used).
Transfection is the process by which DNA is inserted into eukaryotic cells using chemicals or physical forces. It is often easier and less expensive to use chemicals. Protocols vary widely based on the cell type, size of the plasmid, and other conditions. We suggest that you ask around your local university for advice about transfections, or peruse protocols that are provided by transfection kit companies online.
The Mammalian Parts Collection includes several pre-assembled plasmids that express individual transcription units. Each of these plasmids includes a promoter, 5'UTR, Kozak-CDS, 3'UTR and poly(A) tail. Each plasmid encodes different fluorescent proteins (yellow, blue, and red), and their expressions are driven by promoters with varying strengths.
A good way to test transfection efficiency is to transfect a small plasmid that provides a fluorescent readout that can then be measured using a flow cytometer, plate reader, or microscope. The following plasmids in the Collection can be used for this purpose: L1 hEF1a-YFP, L1 PGK-BFP, L1 hEF1a-RFP.
Cells transfected with L1 hEF1a-YFP-IRES-RFP will express both a yellow and red fluorescent protein. This construct encodes both proteins from a single mRNA because it includes an internal ribosome binding site, or IRES.
Plasmids typically don’t replicate in mammalian cells, so the expression levels of a gene within individual cells will naturally fade over time. Therefore, it is necessary, for many applications, to insert a plasmid into the cell’s genome so that it becomes part of the host’s genetic blueprint.
We haven’t included any tools for genome integration in this year’s Mammalian Parts Collection, but perhaps this section will prove useful as a starting point. Briefly, there are four common ways to integrate DNA into the genome: Random integration, transposases, lentiviruses, landing pads, and CRISPR gene-editing tools.
When DNA is transfected into a cell, it sometimes randomly integrates into the genome via a cellular process called non-homologous end joining.
There is also a class of enzymes, called transposases, that randomly integrate DNA fragments into the genome at high efficiency, provided that the DNA is flanked by repeat elements. There are many transposases available, but Sleeping Beauty and PiggyBac are popular choices.
Not all regions of a genome are transcribed as actively as others, either. RNA polymerase enzymes have strong preferences for specific regions of the genome, and so inserting a piece of DNA into a ‘dead’ zone can reduce a gene’s expression levels. PiggyBac tends to insert DNA into more transcriptionally active regions. Large DNA sequences (longer than 6,000 base pairs, for Sleeping Beauty) are usually integrated less efficiently.
A third for genome integration is to use a lentivirus. Briefly, engineered payloads are packaged as RNA into a non-infectious viral vector, and then these viral vectors are used to transduce mammalian cells. The viral vectors enter the cells, reverse transcribe their payloads, and randomly integrate a single copy directly into the genome. A lentivirus can repeat this multiple times in a cell. Sequences packaged within a lentivirus should be less than 10,000 bases in length. Also, if using this method, do not use poly(A) signals in the forward orientation because they will interfere with the virus’ replication.
A fourth strategy for genome integration is to use landing pads, which are DNA sequences pre-inserted into a cell’s genome. Subsequently, different DNA payloads can be inserted directly into the landing pad using an enzyme called a recombinase. Recombinases recognize a specific sequence in the landing pad (e.g., attB for the recombinase, Bxb1) and insert a plasmid containing another specific sequence (attP for Bxb1). Landing pads require up front effort to use, though; they must be integrated into a cell line ahead of time, and they should be placed within transcriptionally active places in the genome. The efficiency of a landing pad integration drops for larger plasmids.
A fifth option – somewhat outside the scope of this Guide – is to use CRISPR-Cas9 gene-editing tools to cleave a specific location within the genome, and then use a repair template to make a site-specific knock-in.
View the full collection of parts in this year's Mammalian Parts Collection on the iGEM website.
Conclusion
You are the future of genetic engineering. This year’s iGEM projects could lay the foundation for an entirely new gene-editing tool, or perhaps devise a method to detect environmental pollutants. Either way, the scientific roots that you grow in the next few months will make a tangible difference in the world. We, at Asimov, want to help you, and we hope this Guide was a useful starting point.
This is a living document. It will continue to expand as we work with iGEM teams, field questions, and discover new biology. Please send feedback and questions to igem2023@asimov.com.
.png)